1. DNA
1.1 DNA basics / structure
DNA (deoxyribonucleic acid) is the genomic material in cells that contains the genetic information used in the development and functioning of all known living organisms. DNA, along with RNA and proteins, is one of the three major macromolecules that are essential for life. Most of the DNA is located in the nucleus, although a small amount can be found in mitochondria (mitochondrial DNA). Within the nucleus of eukaryotic cells, DNA is organized into structures called chromosomes. The complete set of chromosomes in a cell makes up its genome; the human genome has approximately 3 billion base pairs of DNA arranged into 46 chromosomes. The information carried by DNA is held in the sequence of pieces of DNA called genes.
DNA consists of two long polymers of simple units called nucleotides, with backbones made of sugars and phosphate groups joined by ester bonds. These two strands run in opposite directions to each other and are therefore anti-parallel. Attached to each sugar is one of four types of molecules called nucleobases (bases). It is the sequence of these four bases along the backbone that encodes information. The sequence of these bases comprises the genetic code, which subsequently specifies the sequence of the amino acids within proteins. The ends of DNA strands are called the 5′(five prime) and 3′ (three prime) ends. The 5′ end has a terminal phosphate group and the 3′ end a terminal hydroxyl group. One of the major structural differences between DNA and RNA is the sugar, with the 2-deoxyribose in DNA being replaced by ribose in RNA.
 The structure of DNA
The structure of DNA
Bases are classified into two types: the purines, A and G, and the pyrimidines, the six-membered rings C, T and U. Uracil (U), takes the place of thymine in RNA and differs from thymine by lacking a methyl group on its ring. Uracil is not usually found in DNA, occurring only as a breakdown product of cytosine.
In the DNA double helix, each type of base on one strand normally interacts with just one type of base on the other strand. This is complementary base pairing. Therefore, purines form hydrogen bonds to pyrimidines, with A bonding only to T, and C bonding only to G.
1.2 The central dogma
The central dogma of molecular biology is “DNA makes RNA makes protein.” This general rule emphasizes the order of events from transcription through translation and provides the basis for much of the genetic code research in the post double helix 1950s. The central dogma is often expressed as the following: “DNA makes RNA, RNA makes proteins, proteins make us”. Protein is never back translated to RNA or DNA. Furthermore, DNA is never translated directly to protein.
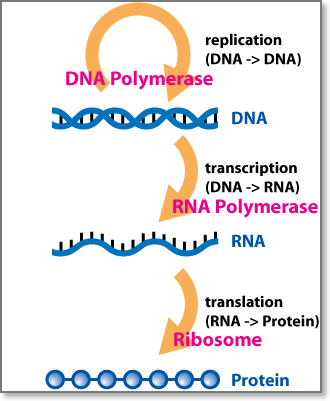 The Central Dogma of Molecular Biology
The Central Dogma of Molecular Biology
See also: The central dogma (external link).
1.3 DNA replication
Cell division is essential for cells to multiply and organisms to grow. As the final step in the Central Dogma, DNA replication must occur in order to faithfully transmit genetic material to the progeny of any cell or organism. When a cell divides, it must correctly replicate the DNA in its genome so that the two daughter cells have the same genetic information as their parent. The double-stranded structure of DNA provides a simple mechanism for DNA replication. The two strands are separated and then an enzyme called DNA polymerase recreates each strand’s complementary DNA sequence. This enzyme makes the complementary strand by finding the correct base through complementary base pairing. As DNA polymerases can only extend a DNA strand in a 5′ to 3′ direction, different mechanisms are used to copy the antiparallel strands of the double helix. In this way, the base on the old strand dictates which base appears on the new strand, and the cell ends up with a perfect copy of its DNA. This process typically takes place during S phase of the cell cycle.
1.4 DNA transcription to RNA
The process by which DNA achieves its control of cell life and function through protein synthesis is called gene expression. A gene is a DNA sequence that contains genetic information for one functional protein. Proteins are essential for the modulation and maintenance of cellular activities. The formation of new protein molecules is made from amino acid building blocks based on information encoded in DNA/RNA. The amino acid sequence of each protein determines its conformation and properties (e.g. ability to interact with other molecules, enzymatic activity etc). Directed protein synthesis follows two major steps: gene transcription and transcript translation.
Transcription is the process by which the genetic information stored in DNA is used to produce a complementary RNA strand. In more detail, the DNA base sequence is first copied into an RNA molecule, called premessenger RNA, by messenger RNA (mRNA) polymerase. Premessenger RNA has a base sequence identical to the DNA coding strand. Genes consist of sequences encoding mRNA (exons) that are interrupted by non-coding sequences of variable length, called introns. Introns are removed and exons joined together before translation begins in a process called mRNA splicing. Messenger RNA splicing has proved to be an important mechanism for greatly increasing the versatility and diversity of expression of a single gene. It takes place in the nucleus in eukaryotes and in the cytoplasm in bacteria and archaea and leads to the formation of mature mRNA. Several different mRNA and protein products can arise from a single gene by selective inclusion or exclusion of individual exons from the mature mRNA products. This phenomenon is called alternative mRNA splicing. It permits a single gene to code for multiple mRNA and protein products with related but distinct structures and functions1. Once introns are excised from the final mature mRNA molecule, this is then exported to the cytoplasm through the nuclear pores where it binds to protein-RNA complexes called ribosomes2. Ribosomes contain two subunits: the 60S subunit contains a single, large (28S) ribosomal RNA molecule complexed with multiple proteins, whereas the RNA component of the 40S subunit is a smaller (18S) ribosomal RNA molecule.
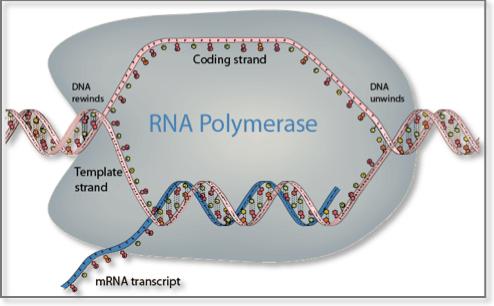 DNA transcription
DNA transcription
1.5 Epigenetics
Although every somatic cell in the human body contains the same genome, activation and silencing of specific genes in a cell-type-specific manner is necessary. Moreover, a cell must silence expression of genes specific to other cell types to ensure genomic stability. This type of repression must be maintained throughout the life of each cell in normal development. Epigenetic modifications that are defined as heritable, yet reversible changes that influence the expression of certain genes but with no alteration in the primary DNA sequence are ideal for regulating these events. The best studied epigenetic modification in human is DNA methylation, however it becomes increasingly acknowledged that DNA methylation does not work alone, but rather occurs in the context of other epigenetic modifications such as the histone modifications.
- DNA methylation: DNA methylation consists of the addition of a methyl group to carbon 5 of the cytosine within the dinucleotide CpG. Regions of DNA in the human genome, ranging from 0.5 to 5 kb, that are CG rich are called CpG islands and are usually found in the promoters of genes. Approximately half of all gene promoters have CpG islands that when methylated lead to transcriptional silencing. De nove DNA methylation is brought about by DNA methyltransferases (DNMT) 3A and 3b whereas DNA methylation maintenance by DNMT1. Aberrant DNA methylation patterns have been described in various human malignancies. In particular, global hupomethylation has been implicated in the earlier stages of carcinogenesis, whereas hypermethylation of tumour suppressor genes has been implicated in cancer progression3. DNA hypomethylating agents are used for the treatment of certain haematological malignancies.
- Histone modifications: Histones are proteins around which DNA winds to form nucleosomes. Nucleosome is the basic unit of DNA packaging within the nucleus and consists of 147 base pairs of genomic DNA wrapped twice around a highly conserved histone octamer, consisting of 2 copies each of the core histones H2A, H2B, H3 and H4. The histone tails may undergo many posttranslational chemical modifications, such as acetylation, methylation, phosphorylation, ubiquitylation, and sumoylation. Combinations of these modifications are thought to constitute a code, named the “histone code”. Histone modifications act except for chromatin condention and transcriptional repression in various other biological processes including gene activation and DNA repair4.
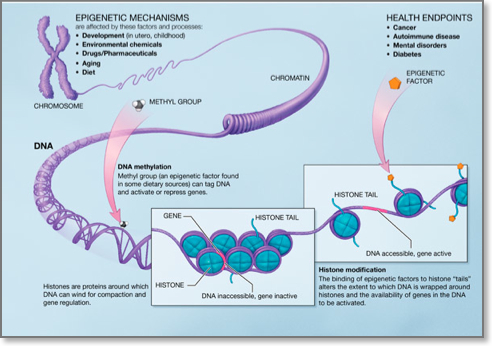 Epigenetic Modifications
Epigenetic Modifications
2. RNA
RNA, is another macromolecule essential for all known forms of life. Like DNA, RNA is made up of nucleotides. Once thought to play ancillary roles, RNAs are now understood to be among a cell’s key regulatory players where they catalyze biological reactions, control and modulate gene expression, sensing and communicating responses to cellular signals, etc.
The chemical structure of RNA is very similar to that of DNA: each nucleotide consists of a nucleobase a ribose sugar, and a phosphate group. There are two differences that distinguish DNA from RNA: (a) RNA contains the sugar ribose, while DNA contains the slightly different sugar deoxyribose (a type of ribose that lacks one oxygen atom), and (b) RNA has the nucleobase uracil while DNA contains thymine. Unlike DNA, most RNA molecules are single-stranded and can adopt very complex three-dimensional structures.
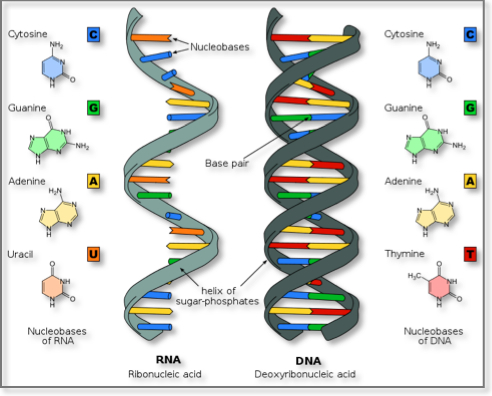 DNA and RNA similarities and differences
DNA and RNA similarities and differences
The universe of protein-coding and non-protein-coding RNAs (ncRNAs) is very diverse vis-à-vis biogenesis, composition and function, and has been expanding rapidly5–9. Among the ncRNAs, microRNAs (miRNAs) represent the best-studied class to date and have been shown to regulate the expression of their protein-coding gene targets in a sequence-dependent manner10–12.
2.1 Monocistronic versus polycistronic RNA
An RNA molecule is said to be monocistronic when it captures the genetic information for a single molecular transcriptional product, e.g. a single miRNA precursor or a single primary mRNA. Most eukaryotic mRNAs are indeed monocistronic. On the other hand, rRNAs and some miRNAs are known to be polycystronic. In the case of polycistronic mRNAs, the primary transcript comprises several back-to-back mRNAs, each of which will be eventually translated into an amino acid sequence (polypeptide). Such polypeptides usually have a related function (they often are the subunits composing a final complex protein) and their coding sequences are grouped into a single primary transcript, which in turn permits them to share a common promoter and to be regulated together.
2.2 Protein-coding RNAs / gene expression
One of the best known and best-studied classes of RNAs are messenger RNAs (mRNAs). MRNAs carry the genetic information that directs the synthesis of proteins by the ribosomes. All cellular organisms use mRNAs. The process of protein synthesis makes use of two more classes of RNAs, the transfer RNAs (tRNAs) and the ribosomal RNAs (rRNAs). The role of tRNAs is the delivery of amino acids to the ribosome where rRNAs link them together to form proteins.
- mRNAs: A fully processed mRNA typically comprises multiple exons that have been assembled into a single chain following splicing of the nascent primary transcript and the removal of intervening introns. The mRNA molecule includes a 5´ cap, the so-called 5´ untranslated region (UTR), the coding region, the 3´UTR, and a variable-length poly(A) tail.
- 5′ cap: The 5´ cap is a modified guanine nucleotide added to the “front” (5´ end) of the pre-mRNA using a 5´-5´-triphosphate linkage. This modification is critical for recognition and proper attachment of mRNA to the ribosome, as well as protection from 5´ exonucleases.
- Untranslated regions: Untranslated regions (UTRs) are nucleotide stretches that flank the coding region and are not translated into amino acids. There are two UTRs: the “five prime untranslated region” or 5´UTR, and the “three prime untranslated region” or 3´UTR. These regions are part of the primary transcript and remain after the splicing of exons into the mRNA. As such UTRs are exonic regions. Several functional roles have been attributed to the untranslated regions, including mRNA stability, mRNA localization, and translational efficiency. The ability and nature of functions performed by a UTR depends on the actual sequence of the UTR and typically differs from one mRNA to the next. The UTRs’ control of translational efficiency has been shown to span the entire spectrum, from enhancement to the complete inhibition of translation. RNA binding proteins that bind to either the 5´ or 3´UTR can influence translation by modulating the ribosome’s ability to bind to the mRNA. Additionally, miRNAs that bind to the 3´UTR may also affect translational efficiency or mRNA stability.
- Coding regions: A subset of the nucleotide sequence that is spanned by the transcript’s exons is used to guide the translation into the corresponding amino acid sequence and is referred to as the coding regions. The length of a coding region is always a multiple of three, and a direct consequence of the fact that each amino acid requires three nucleic acids (the “codon”) for its definition. Since there are 43=64 nucleotide triplets but only 20 amino acids, it follows that a given amino acid can be encoded by more than one triplets. The correspondence between a triplet and an amino acid is given by the codon table which also defines the ‘genetic code.’ The codon tables of organisms are largely identical but slight variations have been discovered over the course of the last 30 years. Codons are ‘decoded’ and translated into peptide polymers by the ribosome. Coding regions begin with the start codon and end with a stop codon. In general, the start codon is an AUG triplet and the stop codon is one of UAA, UAG, or UGA.
- Poly(A) tail: The variable length 3´ poly(A) tail is a long sequence of adenine nucleotides (often several hundred) added to the 3´ end of the pre-mRNA. This tail promotes export from the nucleus, translation, and stability of mRNA13,14.
 The structure of an mRNA
The structure of an mRNA
3. RNA Interference
RNA interference is a process that moderates gene expression in a sequence dependent manner. The RNAi pathway is found in all higher eukaryotes and was recently found in the budding yeast as well. Viruses have also been shown to be RNAi-aware in that they use their natural host’s RNAi pathway to their benefit.
RNAi is initiated by Dicer, a double-stranded-RNA-specific endonuclease from the RNase III protein family. Dicer cleaves double-stranded RNA (dsRNA) molecules into short fragments of ~21 nucleotides, with a two-nucleotide overhang at their 3′ end, as well as a 5′ phosphate and a 3′ hydroxyl group. The RNAi pathway can be engaged by two types of small regulatory non-coding RNAs: a) small interfering RNAs (siRNAs), which are typically exogenous, and b) microRNAs (miRNAs), which are endogenous. SiRNAs are double-stranded ncRNAs that are mainly delivered to the cell experimentally by various transfection methods although they have been described to be produced form the cell itself15. MiRNAs are another type of small ncRNAs that are transcribed from the organism’s DNA. After processing of the primary siRNAs and miRNAs by Dicer, typically one of the two strands is loaded onto the RNA-induced silencing complex (RISC), a complex of RNA and proteins that includes the Argonaute protein, whereas the other strand is discarded. The loaded siRNAs and miRNAs guide RISC’s binding to specific mRNAs (targets). The sequence of the siRNA/miRNA determines the identity of the target. The resulting heteroduplex of the siRNA/miRNA and its target mRNA is characterized by base-pairing that generally spans much of the siRNA/miRNA’s length. SiRNAs are typically designed to be perfectly complementary to their targets. On the other hand, miRNAs need not be fully-complementary to the mRNA that they target. This imprecise matching gives miRNAs the potential to target multiple endogenous mRNAs simultaneously. Whether induced by an siRNA or an miRNA, the downstream effect is the down-regulation of the targeted mRNA either via degradation or translational inhibition.
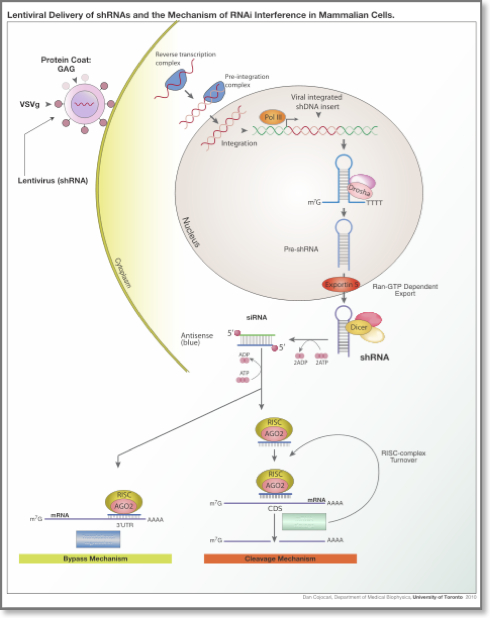 RNA interference in mammalian cells
RNA interference in mammalian cells
Designer siRNAs are now widely used in the laboratory to down-regulate specific proteins whose function is under study. At the same time, the ability to engage the RNAi pathway in an on demand manner suggests the possibility that RNAi can be used in the clinic to reduce the production of those proteins that are over-expressed in a given disease context. Analogously, RNAi can also be used to “sponge” away excess amounts of an endogenous miRNA that would otherwise down-regulate a needed protein. The delivery method remains an important consideration for the development of RNAi-based therapies as the active molecule needs to be delivered efficiently and in a tissue-specific manner in order to maximize impact and diminish off-target effects.
See also: RNAi (external link).
4. Non protein coding RNAs (a.k.a non-coding RNAs or ncRNAs)
The expression of proteins is determined by genomic information, and their presence supports the function of cell life. Parts of an organism’s genome are transcribed in an orderly tissue- and developmental phase- specific manner into RNA transcripts that are destined to effect the eventual production of proteins.
Until fairly recently, it was believed that the molecules that are important for the function of a cell are those described by the “Central Dogma” of biology, namely messenger RNAs and proteins. Things began to change with the discovery of microRNAs more than 20 years ago in plants16 and animals17,18. Subsequent research efforts have demonstrated that large parts of an organism’s genome will be transcribed at one time point or another into RNA, but will not be translated into an amino acid sequence. These RNA transcripts have been referred to as ncRNAs and there is increased appreciation that many of them are indeed functional and affect key cellular processes.
There are many recognizable classes of ncRNAs, each having a distinct functionality. These include: transfer RNAs (tRNAs)19; ribosomal RNAs (rRNAs)20; the above-mentioned miRNAs17,18; small nucleolar RNAs (snoRNAs)21,22; piwi-interacting (piRNAs)23–25; transcription initiation RNAs (tiRNAs)26; human microRNA-offset (moRNAs)27; sno-derived RNAs (sdRNAs)28; long intergenic ncRNAs (lincRNAs)29; etc. The full extent of distinct classes of ncRNAs that are encoded within the human genome is currently unknown but are believed to be numerous.
- Short non-coding RNAs: At least three classes of small RNAs are encoded in our genome, based on their biogenesis mechanism and the type of Ago protein that they are associated with miRNAs, endogenous siRNAs and piRNAs. It should be noted, however, that the recent discoveries of numerous non‐canonical small RNAs have somewhat blurred the boundaries between the classes.
- MicroRNAs (miRNAs): MicroRNAs (miRNAs) comprise a large family of naturally occurring, endogenous, single-stranded ~22-nucleotide-long RNAs. MiRNAs function as key post-transcriptional regulators of gene expression by base-pairing with their target mRNAs. Originally believed to effect their impact exclusively through the target mRNAs 3´UTR30, they have since been shown to have extensive coding region targets as well12,31. More than one thousand miRNAs are currently known for the human genome, and each of them has the ability to down regulate the expression of possibly thousands of protein coding genes32. In mammals, miRNAs are predicted to control more than ~90% of all protein-coding genes32.
- miRNA biogenesis: MicroRNAs (miRNAs) are either transcribed explicitly by RNA polymerase II or generated from the appropriately-long introns of protein-coding genes as a by-product of splicing.
Canonical Pathway
- In the canonical pathway, transcription of the primary miRNA precursor (pri-miRNA) is carried out by RNA polymerase II. The pri-miRNA is processed into a precursor miRNA (pre-miRNA) by the “microprocessor complex” which comprises Drosha, a member of the RNase III family of endonucleases, and DGCR8, a double-stranded-RNA-binding protein. Pre-miRNAs are generaly 60-70-nucleotides in length, have a two-nucleotide overhang at the 3′ end and a 5′ phosphate group, and fold into a characteristic hairpin-like structure. Exportin-5 recognizes the two-nucleotide 3´-overhang, characteristic of RNase III-mediated cleavage, and shuttles the pre-miRNA through the nuclear pore into the cytoplasm, where it is further processed by Dicer, another endonuclease. Dicer pairs with TRBP and PACT, both double-stranded-RNA-binding proteins, and cleaves the pre-miRNA to form a transient ~22-nucleotide double stranded RNA, again with two-nucleotide overhangs at the 3´end. One of the two strands, typically the one with a relatively lower stability of base-pairing at the 5´-end (“the thermodynamic asymmetry rule”) is referred to as the “guide” strand and gives rise to the “mature” miRNA that associates with the Argonaute (AGO) protein to form the core of miRNA-associated RISC (miRISC) or simply, RISC. The second strand, referred to as the “passenger” strand is typically degraded. However, there have been well-documented examples of passenger strands giving rise to mature products, known as the miRNA*, which are also involved in regulatory activities. The miRNA helps direct RISC to targets in a sequence-dependent manner thereby mediating the repression of the target’s expression.
Alternative pathways (non-canonical)
- Drosha independent pathways: As mentioned above, most miRNAs either originate form their own transcription units or derive from the exons or introns of other genes33 and require both Drosha and Dicer for cleavage in their maturation. It was recently shown however first in Droshophila33 and later in mammals34 that short hairpin introns, called mirtrons can be alternative sources of miRNAs. Although there are several differences between mammalian and invertebrate mirtrons, both are Drosha independent. Mirtrons are short introns with hairpin potential that can be spliced and debranched into pre-miRNA mimics and then enter the canonical pathway. Post nuclear export, they can then be cleaved by Dicer and incorporated into RISC34.
- Dicer independent pathways: MiRNA biogenesis independent of Dicer has only been described thus far for miR-45134. This miRNA is processed by Drosha but its does not require Dicer. Instead, its pre-miRNA, once loaded into Ago, is cleaved by the Ago catalytic centre to generate an intermediate 3’ end, which is further trimmed. Importantly, the Ago catalytic function for the miR-451 biogenesis was shown in Ago2 homozygous mutants that were found to have loss of miR-451 and died shortly after their birth with anemia34.
 miRNA biogenesis
miRNA biogenesis - miRNA function: For miRNAs to pair with their target mRNAs, a region of the miRNA sequence near its 5′ end needs to be involved in base-pairing. The region in question spans nucleotide positions 2 through 7 inclusive and is known as the ‘seed’. Not all nucleotides of the seed region need to be paired for the heteroduplex to have a functional effect18,35–37. The base-pairing in the seed region can comprise Watson-Crick bonding, although this was recently shown to neither be necessary31 nor sufficient37.In animals, it was believed for many years that the miRNA-binding sites are exclusively in the 3′UTR of mRNAs. However, it was recently shown that animal miRNAs could target mRNA coding regions equally effectively and extensively31. In plants, miRNA targeting is predominantly through coding region targets.The ways in which miRNAs cause down-regulation of their target mRNAs has been hotly debated. The possible mechanisms include: translational inhibition38; removal of the poly(A) tail from mRNAs (deadenylation)39,40; disruption of cap–tail interactions41,42; and, mRNA degradation by exonucleases43,44, although highly complementary targets can be cleaved endonucleolytically45. Other types of regulatory function of miRNAs have also been described, and include translational activation46, heterochromatin formation47, and DNA methylation48.
- miRNA nomeclature: Each miRNA is identified by a unique numerical name. The standard naming system uses abbreviated three letter prefixes to designate the species (e.g., hsa- in Homo sapiens, which is usually ignored in the literature when the organism is implied), followed by the three-letter tag mir or miR, followed by a number. The number is assigned by the miRBase Registry. The mature sequences are designated using the tag ‘miR’ (capitalized R), whereas the precursor hairpins use the tag ‘mir’. Orthologous miRNAs across organisms differ only in their species name (e.g., hsa-miR-101 in humans vs. mmu-miR-101 in mice). Nearly identical miRNAs that differ at only one or two positions are distinguished by lettered suffixes (e.g., miR-10a and miR-10b). Paralogous miRNAs, i.e. miRNAs whose precursors have multiple instances, i.e. distinct loci, in the same genome are indicated by numbered suffixes (e.g., mir-281-1 and mir-281-2); such precursors give rise to identical mature miRNAs, but not necessarily with the same time kinetics.
- Ultraconserved genes or UCGs: UCGs are ncRNAs that are transcribed from ultraconserved regions (UCRs)8,49. UCRs are genomic segments that have identified to be 100% conserved between orthologous regions in the human, mouse and rat. Of these, 481 are transcribed (T-UCRs) from integenic sequences (39%), from introns (43%) while the remainder is exonic or exon overlapping50,51.
- Small nucleolar RNAs (snoRNAs): Small nucleolar RNAs (snoRNAs) are a highly evolutionarily conserved class of RNAs and have been considered on of the best-characterized classes of nc-RNAs22. They are intermediate-sized RNAs of 60-300 nucleotides in length and are predominantly found in the nucleus52. Two major classes of snoRNAs have been identified which possess distinctive, evolutionary conserved sequence elements. One group contains the the box C/D motif, whereas snoRNAs in the other group carry the box H/ACA elements53. SnoRNAs are components of the small nucleolar ribonucleoproteins (snoRNPs) and are involved in the post-transcriptional modification of rRNAs and some splicesomal RNAs52,54,55. In particular, most C/D and H/ACA snoRNAs function in 2’-O-methylation and pseudouridylation respectively of various classes of RNAs. These modifications are important for the production of efficient ribosomes56. Recently, many “orphan” snoRNAs that lack complementarities to rRNAs, tRNAs or other known stable RNAs have been identified57, suggesting that they might function in cellular processes other than RNA modification.
- piwi-interacting (piRNAs): PIWI-interacting RNAs (piRNAs) are 25–30 nucleotides in length and have been found in most metazoans. They are Dicer-independent and they bind to particular Argonaute proteins called PIWI proteins24. These RNA-protein complexes are involved in the epigenetic and and post-transcriptional gene silencing of transposable and other repetitive elements58,59. PiRNAs have also been recently linked to the regulation of imprinted-DNA methylation60.
- transcription initiation RNAs (tiRNAs): Transcription initiation RNAs (tiRNAs) are derived from sequences on the same strand as the transcription start sites (TSS) and are preferentially associated with GC rich promoters. They have been found to have a modal lenth of 18 nucleotides that map within -60 and +120 nucleotides of TSS. They are associated with highly expressed transcripts and sites of RNA polymerase II binding26.
- human microRNA-offset (moRNAs): MicroRNA-offset RNAs (moRNAs) are generated from sequences immediately adjacent to to mature miR and miR* loci. They have been found in the tunicate Ciona intestinalis but also in human microRNA precursors, albeit in low levels27,61. The high level of conservation and the example of miR-219 with moRNAs conserved between humans and Ciona suggests that they might have a functional role27,61.
- sno-derived RNAs (sdRNAs): Sno-derived RNAs (sdRNAs) comprise a novel class of small RNAs in eykaryotes. The ones that derive from H/ACA snoRNAs are predominantly 20–24 nucleotides long and originate from the 3’ end, where as those derived from C/D snoRNAs are either 17–19 nt long or >27 nt long and predominantly originate from the 5’ end. 28 Through a comparison of human small RNA deep sequencing data sets it was shown that box C/D sdRNA accumulation patterns are conserved across multiple cell types, although the ratio of the abundance of different sdRNAs from a given snoRNA varied62.
- Long non-coding RNAs: Long non-coding RNAs are a heterogenous group of non-coding transcripts that are longer than 200 nucleotides (a rather arbitrarily condition/limit that ignores the possibility of ncRNAs with lengths between 40 and 200) that are involved in various processes. A large number of such RNAs have been identified and constitute the largest portion of the mammalian non-coding transcriptome. Such RNAs have been identified in both protein-coding loci and also within intergenic stretches. Numerous protein-coding loci give rise to non-protein-coding RNA , with notable examples being β-actin, γ-actin, RB1 etc.
- Other categories: Long intergenic RNAs (lincRNAs), and others with enhancer-like functions were described only recently29,63. Attempts to functionalize these other classes of ncRNAs are currently in their very early stages. LincRNAs arise from intergenic regions and exhibit a specific chromatin signature that consists of a short stretch of trimethylation of histone protein H3 at the lysine in position 4 (H3K4me3) – characteristic of promoter regions, followed by a longer stretch of trimethylation of histone H3 at the lysine in position 36 (H3K36me3) – characteristic of transcribed regions. This lincRNA profile is also known as “K4-K36 signature”. Transcripts from active enhancer regions with another chromatin signature, the H3 lysine 4 monomethylation (H3K4me1) modification have also been described, although it is not clear whether they represent a distinct class of lincRNAs.
The biological role of long ncRNAs as a class remains largely elusive. Several specific cases have been shown to be involved in transcriptional gene silencing, and the activation of critical regulators of development and differentiation: these exerted their regulatory roles by interfering with transcription factors or their co-activators, though direct action on DNA duplex, by regulating adjacent protein-coding gene expression, by mediating DNA epigenetic modifications, etc.
4.1 RNA splicing
is a complex process mediated by a large RNA-containing protein called a spliceosome. This consists of five types of small nuclear RNA molecules (snRNA) and more than 50 proteins (small nuclear riboprotein particles).4.2 RNA reverse transcription
Reverse transcription is the transfer of information from RNA to DNA (the reverse of normal transcription). This is known to occur in the case of retroviruses, such as HIV, as well as in eukaryotes, in the case of retrotransposons and telomere synthesis.
4.3 RNA editing / post-transcriptional modifications
Post-transcriptional modification is a process in cell biology by which, primary transcript RNA is converted into mature RNA. A notable example is the conversion of precursor messenger RNA into mature messenger RNA (mRNA), which includes splicing and occurs prior to protein synthesis. This process is vital for the correct translation of the genomes of eukaryotes as the human primary RNA transcript that is produced as a result of transcription contains both exons, which are coding sections of the primary RNA transcript and introns, which are the non coding sections of the primary RNA transcript.
Post-trancriptional modifications that lead to a mature mRNA include the (i) addition of a methylated guanine cap to the 5′ end of mRNA and (ii) the addition of a poly-A tail to the other end. The cap and tail protect the mRNA from enzyme degradation and aid its attachment to the ribosome. In addition, (iii) introns (non-coding) sequences are spliced out of the mRNA and exons (coding) sequences are spliced together. The mature mRNA transcript will then undergo translation64.
5. Proteins
A protein is a molecule that performs reactions necessary to sustain the life of an organism. One cell can contain thousands of proteins.
5.1 RNA translation
Following transcription, translation is the next step of protein biosynthesis. In translation, mRNA produced by transcription is decoded by the ribosome to produce a specific amino acid chain, or a polypeptide, that will later fold into a protein. Ribosomes read mRNA sequence in a ticker tape fashion three bases at a time, inserting the appropriate amino acid encoded by each three-base code word or codon into the appropriate position of the growing protein chain. This process is called mRNA translation. In particular, the mRNA sequence directly relates to the polypeptide sequence by binding to transfer RNA (tRNA) adapter molecules in binding pockets within the ribosome. Each amino acid is encoded by a sequence of three successive bases. Because there are four code letters (A, C, G, and U), and because sequences read in the 5′ → 3′ direction have a different biologic meaning than sequences read in the 3′ → 5′ direction, there are 43=64, possible codons consisting of three bases. Some specialized codons serve as punctuation points during translation. The methionine codon (AUG), serves as the initiator codon signaling the first amino acid to be incorporated. All proteins thus begin with a methionine residue, but this is often removed later in the translational process. Three codons, UAG, UAA, and UGA, serve as translation terminators, signaling the end of translation. The completed polypeptide chain then folds into a functional three-dimensional protein molecule and is transferred to other organelles for further processing or released into cytosol for association of the newly completed chain with other subunits to form complex multimeric proteins.
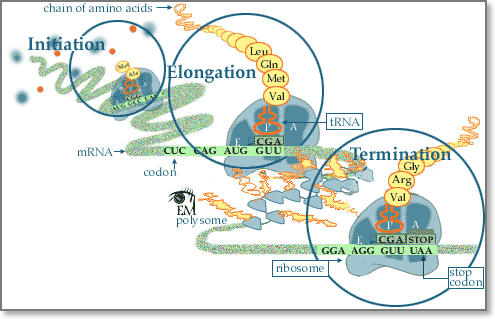 Protein translation
Protein translation
5.2 Post-translational modifications
Post-translational modification is the chemical modification of a peptide that takes place after its translation. They represent one of the later steps in protein biosynthesis for many proteins. During protein synthesis, 20 different amino acids can be incorporated in order to form a polypeptide. After translation, the addition of other biochemical functional groups (such as acetate, phosphate, various lipids and carbohydrates) to the protein’s amino acids extends the range of functions of the protein modifying the chemical nature of an amino acid (e.g. citrullination), or making structural changes (e.g. formation of disulfide bridges). In addition, enzymes may remove amino acids from the amino end of the protein, or even cut the peptide chain in the middle. For instance, most nascent polypeptides start with the amino acid methionine because the “start” codon on mRNA also codes for this amino acid. This amino acid is usually taken off during post-translational modification. Other modifications, like phosphorylation, are part of common mechanisms for controlling the behavior of a protein, for instance activating or inactivating an enzyme.
See also: Inside a cell (external link).
References
- Bentley D. The mRNA assembly line: transcription and processing machines in the same factory. Current opinion in cell biology. 2002; 14(3): 336-42
- Lee TI, Young RA. Transcription of eukaryotic protein-coding genes. Annual review of genetics. 2000; 34: 77-137
- Esteller M. Epigenetics in cancer. The New England journal of medicine. 2008; 358(11): 1148-59
- Kouzarides T. Chromatin modifications and their function. Cell. 2007; 128(4): 693-705
- Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006; 15 Spec No 1: R17-29
- Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004; 306(5705): 2242-6
- Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005; 308(5725): 1149-54
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, Maeda N, et al. The transcriptional landscape of the mammalian genome. Science. 2005; 309(5740): 1559-63
- Khaitovich P, Kelso J, Franz H, Visagie J, Giger T, Joerchel S, et al. Functionality of intergenic transcription: an evolutionary comparison. PLoS Genet. 2006; 2(10): e171
- Ambros V. The evolution of our thinking about microRNAs. Nat Med. 2008; 14(10): 1036-40
- Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009; 136(2): 215-33
- Rigoutsos I. New tricks for animal microRNAS: targeting of amino acid coding regions at conserved and nonconserved sites. Cancer research. 2009; 69(8): 3245-8
- Bernstein P, Ross J. Poly(A), poly(A) binding protein and the regulation of mRNA stability. Trends in biochemical sciences. 1989; 14(9): 373-7
- Guhaniyogi J, Brewer G. Regulation of mRNA stability in mammalian cells. Gene. 2001; 265(1-2): 11-23
- Watanabe T, Totoki Y, Toyoda A, Kaneda M, Kuramochi-Miyagawa S, Obata Y, et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature. 2008; 453(7194): 539-43
- Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science. 1999; 286(5441): 950-2
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993; 75(5): 843-54
- Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993; 75(5): 855-62
- Eddy SR. Non-coding RNA genes and the modern RNA world. Nature reviews Genetics. 2001; 2(12): 919-29
- Smit S, Widmann J, Knight R. Evolutionary rates vary among rRNA structural elements. Nucleic acids research. 2007; 35(10): 3339-54
- Huttenhofer A, Brosius J, Bachellerie JP. RNomics: identification and function of small, non-messenger RNAs. Current opinion in chemical biology. 2002; 6(6): 835-43
- Bachellerie JP, Cavaille J, Huttenhofer A. The expanding snoRNA world. Biochimie. 2002; 84(8): 775-90
- Aravin A, Gaidatzis D, Pfeffer S, Lagos-Quintana M, Landgraf P, Iovino N, et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature. 2006; 442(7099): 203-7
- Girard A, Sachidanandam R, Hannon GJ, Carmell MA. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature. 2006; 442(7099): 199-202
- Grivna ST, Beyret E, Wang Z, Lin H. A novel class of small RNAs in mouse spermatogenic cells. Genes & development. 2006; 20(13): 1709-14
- Taft RJ, Glazov EA, Cloonan N, Simons C, Stephen S, Faulkner GJ, et al. Tiny RNAs associated with transcription start sites in animals. Nature genetics. 2009; 41(5): 572-8
- Langenberger D, Bermudez-Santana C, Hertel J, Hoffmann S, Khaitovich P, Stadler PF. Evidence for human microRNA-offset RNAs in small RNA sequencing data. Bioinformatics. 2009; 25(18): 2298-301
- Taft RJ, Glazov EA, Lassmann T, Hayashizaki Y, Carninci P, Mattick JS. Small RNAs derived from snoRNAs. RNA. 2009; 15(7): 1233-40
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009; 458(7235): 223-7
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116(2): 281-97
- Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I. MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature. 2008; 455(7216): 1124-8
- Miranda KC, Huynh T, Tay Y, Ang YS, Tam WL, Thomson AM, et al. A pattern-based method for the identification of MicroRNA binding sites and their corresponding heteroduplexes. Cell. 2006; 126(6): 1203-17
- Du T, Zamore PD. microPrimer: the biogenesis and function of microRNA. Development. 2005; 132(21): 4645-52
- Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature. 2010; 465(7298): 584-9
- Ha I, Wightman B, Ruvkun G. A bulged lin-4/lin-14 RNA duplex is sufficient for Caenorhabditis elegans lin-14 temporal gradient formation. Genes & development. 1996; 10(23): 3041-50
- Vella MC, Choi EY, Lin SY, Reinert K, Slack FJ. The C. elegans microRNA let-7 binds to imperfect let-7 complementary sites from the lin-41 3’UTR. Genes & development. 2004; 18(2): 132-7
- Didiano D, Hobert O. Perfect seed pairing is not a generally reliable predictor for miRNA-target interactions. Nature structural & molecular biology. 2006; 13(9): 849-51
- Djuranovic S, Nahvi A, Green R. miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science. 2012; 336(6078): 237-40
- Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annual review of biochemistry. 2010; 79: 351-79
- Wu L, Fan J, Belasco JG. MicroRNAs direct rapid deadenylation of mRNA. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103(11): 4034-9
- Eulalio A, Rehwinkel J, Stricker M, Huntzinger E, Yang SF, Doerks T, et al. Target-specific requirements for enhancers of decapping in miRNA-mediated gene silencing. Genes & development. 2007; 21(20): 2558-70
- Wang B, Love TM, Call ME, Doench JG, Novina CD. Recapitulation of short RNA-directed translational gene silencing in vitro. Molecular cell. 2006; 22(4): 553-60
- Ramachandran V, Chen X. Degradation of microRNAs by a family of exoribonucleases in Arabidopsis. Science. 2008; 321(5895): 1490-2
- Chatterjee S, Grosshans H. Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature. 2009; 461(7263): 546-9
- Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004; 304(5670): 594-6
- Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nature reviews Genetics. 2008; 9(2): 102-14
- Kim DH, Saetrom P, Snove O, Jr., Rossi JJ. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105(42): 16230-5
- Tan Y, Zhang B, Wu T, Skogerbo G, Zhu X, Guo X, et al. Transcriptional inhibiton of Hoxd4 expression by miRNA-10a in human breast cancer cells. BMC molecular biology. 2009; 10: 12
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, et al. Ultraconserved elements in the human genome. Science. 2004; 304(5675): 1321-5
- Calin GA, Liu CG, Ferracin M, Hyslop T, Spizzo R, Sevignani C, et al. Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer cell. 2007; 12(3): 215-29
- Mestdagh P, Fredlund E, Pattyn F, Rihani A, Van Maerken T, Vermeulen J, et al. An integrative genomics screen uncovers ncRNA T-UCR functions in neuroblastoma tumours. Oncogene. 2010; 29(24): 3583-92
- Tollervey D, Kiss T. Function and synthesis of small nucleolar RNAs. Current opinion in cell biology. 1997; 9(3): 337-42
- Kiss T. Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell. 2002; 109(2): 145-8
- Kiss-Laszlo Z, Henry Y, Bachellerie JP, Caizergues-Ferrer M, Kiss T. Site-specific ribose methylation of preribosomal RNA: a novel function for small nucleolar RNAs. Cell. 1996; 85(7): 1077-88
- Ni J, Tien AL, Fournier MJ. Small nucleolar RNAs direct site-specific synthesis of pseudouridine in ribosomal RNA. Cell. 1997; 89(4): 565-73
- Decatur WA, Fournier MJ. rRNA modifications and ribosome function. Trends in biochemical sciences. 2002; 27(7): 344-51
- Huttenhofer A, Kiefmann M, Meier-Ewert S, O’Brien J, Lehrach H, Bachellerie JP, et al. RNomics: an experimental approach that identifies 201 candidates for novel, small, non-messenger RNAs in mouse. The EMBO journal. 2001; 20(11): 2943-53
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, et al. A slicer-mediated mechanism for repeat-associated siRNA 5′ end formation in Drosophila. Science. 2007; 315(5818): 1587-90
- Pal-Bhadra M, Leibovitch BA, Gandhi SG, Rao M, Bhadra U, Birchler JA, et al. Heterochromatic silencing and HP1 localization in Drosophila are dependent on the RNAi machinery. Science. 2004; 303(5658): 669-72
- Watanabe T, Tomizawa S, Mitsuya K, Totoki Y, Yamamoto Y, Kuramochi-Miyagawa S, et al. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science. 2011; 332(6031): 848-52
- Shi W, Hendrix D, Levine M, Haley B. A distinct class of small RNAs arises from pre-miRNA-proximal regions in a simple chordate. Nature structural & molecular biology. 2009; 16(2): 183-9
- Scott MS, Ono M, Yamada K, Endo A, Barton GJ, Lamond AI. Human box C/D snoRNA processing conservation across multiple cell types. Nucleic acids research. 2012; 40(8): 3676-88
- Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, et al. Long noncoding RNAs with enhancer-like function in human cells. Cell. 2010; 143(1): 46-58
- Lodish HF BA, Kaiser C, Krieger M, Scott MP, Bretscher A, Ploegh H, Matsudaira PT. Chapter 8: Post-transcriptional Gene Control. In: Freeman W, editor. Molecular Cell Biology. San Francisco; 2007