1. Polymerase chain reaction (PCR)
The development of the polymerase chain reaction was a major breakthrough that has revolutionized the utility of a genome-based strategy for diagnosis and treatment of disease. It permits the detection, synthesis, and isolation of specific genes and allows for the differentiation of alleles of a gene that differ by as little as one nucleotide. It does not require sophisticated equipment or unusual technical skills. A clinical specimen consisting of only minute amounts of tissue is sufficient. Additionally, in most circumstances, no special preparation of the tissue is necessary. Thus, the polymerase chain reaction makes recombinant DNA techniques accessible to clinical laboratories. This single advance has resulted in a tremendous increase in the use of direct gene analysis for the diagnosis of human diseases.
DNA is a double stranded molecule, with two complementary strands being held together by the rules of Watson-Crick base pairing. In terms of orientation, the two strands are antiparallel to one another. The polymerase chain reaction exploits this double stranded structure and the naturally occurring process of copying an existing DNA strand by DNA polymerase. The required ingredients comprise an existing denatured strand of DNA, which is used as the template, and a primer. Primers are short oligonucleotides, 12 to 100 bases in length, with sequence that is complementary to the desired region of the existing DNA strand. The primer effectively “tells” the polymerase enzyme “where” to begin the process of copying. If the base sequence of the DNA of the gene under study is known, two synthetic oligonucleotides complementary to sequences flanking the region of interest can be prepared. If these are the only oligonucleotides present in the reaction mixture, then the DNA polymerase can only generate, i.e. “copy”, daughter strands of DNA downstream from those oligonucleotides that are identical to the target DNA. At the end of one such cycle, the net effect is synthesis of one more copy for each instance of the DNA sequence bounded by the two used primers: this effectively doubles the number of copies of the DNA region of interest with each cycle. Subsequent denaturation of DNA by heat allows hybridization (annealing) of the now-single-stranded daughter strands to the available primers in preparation for elongation and the completion of one more cycle. Each repetition increases the original number of available copies of the region of interest by 2x, 4x, 8x, 16x, 32x, 64x etc. Thirty executions of the cycle result in an increase of the original copy number by more than a billion times. PCR results in the selective amplification of a region of interest that can yield micrograms of product from a significantly smaller initial amount.
To determine whether the PCR generated the targeted DNA fragment (sometimes referred to as the amplicon), agarose gel electrophoresis is employed for size separation of the PCR products. The sizes of the PCR products are determined by comparison with a DNA ladder (a molecular-weight marker), which contains DNA fragments of known size, and is run on the gel alongside the PCR products.
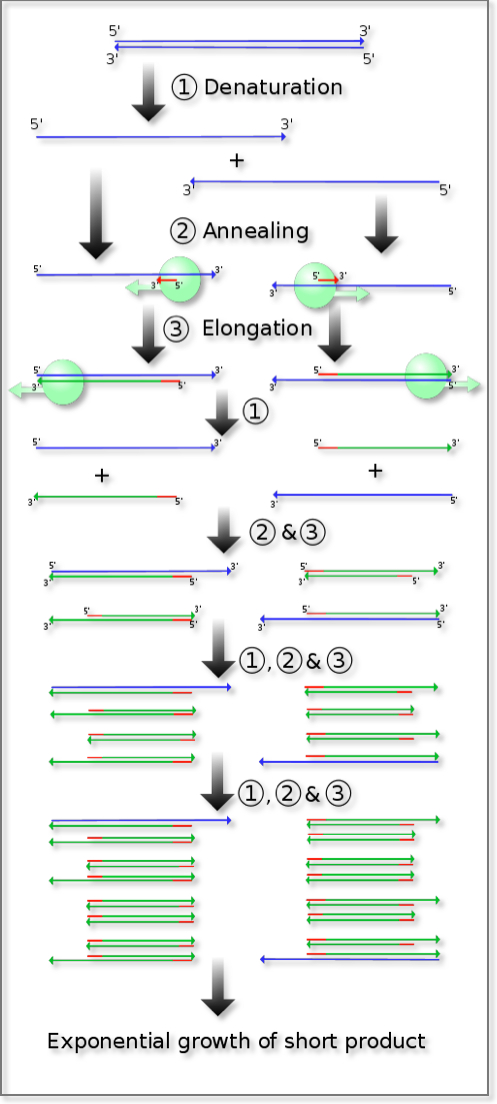
PCR
1.1 Real time PCR
Quantitative PCR (Q-PCR) is also called real-time PCR and is a technique based on the PCR that is used to simultaneously amplify and quantify the targeted DNA region or targeted molecule (referred to as the PCR product). The technique can quantitatively measure amounts of DNA, RNA and cDNA in the reaction.
The procedure follows the same basic principle as PCR. Its main feature is that the amplified DNA product is detected as the reaction progresses in real time, hence the name. The most common methods used for detection of products in real-time PCR are: (1) non-specific fluorescent dyes that bind to any double-stranded DNA (e.g. SYBR green), and (2) sequence-specific DNA probes consisting of oligonucleotides that are labeled with a fluorescent reporter. Detection is possible only after hybridization of the probe with its complementary DNA target.
PCR can also be employed to robustly detect and quantify the expression of transcripts (both protein-coding and non-coding) from small amounts of RNA. Amplification of the gene transcript is again necessary, however the RNA sample is first reverse transcribed to cDNA with reverse transcriptase (see RT-PCR). Combination of PCR techniques based on reverse transcription and fluorophores permits measurement of cDNA amplification during PCR in real time, i.e., the amplified product is measured at each PCR cycle. The data generated can be further analyzed by computer software to calculate relative gene expression in several samples, or mRNA copy number.
1.2 RT-PCR
Reverse transcription polymerase chain reaction (RT-PCR) is a variant of PCR. It is a technique commonly used where a RNA strand is reverse transcribed into its DNA complement (complementary DNA, or cDNA) using the enzyme reverse transcriptase, and the resulting cDNA is amplified using PCR. Reverse transcription PCR is abbreviated as RT-PCR but should not be confused with real-time polymerase chain reaction which is abbreviated as Q-PCR or qRT-PCR.
2. Microarrays
A microarray consists of a small membrane or glass slide containing probes corresponding to genomic regions of interest and arranged in a regular, matrix-like pattern. It provides a mechanism for interrogating en mass samples of unknown composition for the presence or absence of regions of interest (represented by each of the probes). Each DNA spot contains a few picomoles of the specific DNA sequence from the region of interest. The microarray approach relies on Watson-Crick base pairing rules and the existence of reverse complementarity between the probe/bait/feature and its sought molecule.
A higher number of complementary base pairs results in stronger hybridization (non-covalent bonding) between the probe and its target. The resulting strength is determined by the number of paired bases, the hybridization conditions (such as temperature) and on washing after hybridization. Total strength of the signal from a given spot of the microarray matrix depends upon the amount of target sample binding to the probes present at that spot. Microarrays use relative quantization in which the intensity of a feature is compared to the intensity of the same feature under a different condition. Lastly, the identity of a bait/probe/feature is known by virtue of knowing its position on the array.
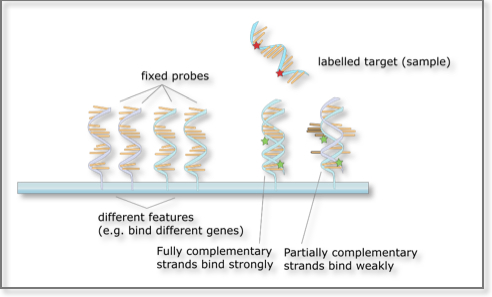 Microarrays: The basic principle
Microarrays: The basic principle
There are two major applications of the DNA microarray technology: 1) identification of a targeted sequence (gene, gene mutation, transcript of interest); and 2) determination of the expression level, i.e. abundance of the targeted sequence. In the latter case, the RNA to be queried is first reverse-transcribed into cDNA which is then allowed to hybridize with the microarray probes. The process of measuring gene expression via cDNA is called expression analysis or expression profiling. Expression profiling can be done within a single sample, between two different cell types or tissue samples, such as in healthy and cancer tissue, etc.
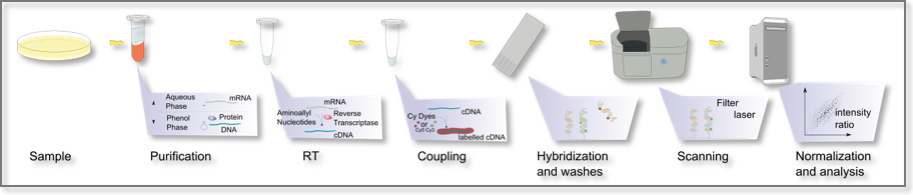 Microarrays: A typical experiment
Microarrays: A typical experiment
Microarrays represent a significant advance both because of their small size and the very large number of probes (tens of thousands to hundreds of thousands) they can contain. Not surprisingly, they have dramatically accelerated many types of investigation.
3. Transfection
Transfection is the process of deliberately introducing a purified segment of nucleic acids into mammalian cells. Protein-coding RNA but also plasmid DNA or siRNA (silencing RNA) can be transfected into cells to transiently express the corresponding product. One particular variant, transfection with siRNAs has become a major application in research. See also RNAi – internal link.
Transfection can be stable or transient. Stable transfection occurs after the transfected material becomes incorporated into the recipients genome where it can be replicated and expressed along with the host genome. In contrast, if the host cell rejects the inserted genomic segment, the transfection is transient because expression of the introduced genetic material is soon lost, lasting only a short duration of time. Transient transfection experiments are less complex than stable transfection ones. Stable and transient transfection processes offer varying benefits and are used in different application settings.
There are various methods of introducing nucleic acid molecules into an eukaryotic cell. They can be chemistry based or they can rely on physical treatment. Alternatively, biological particles can be used as carriers. For most applications, it is sufficient that the genetic material is only transiently expressed. The foreign DNA will be diluted through mitosis since it is not usually integrated into the nuclear genome. If however the transfected gene is desired to remain, then a stable transfection must occur. To select a stably transfected cell, a selection marker is co-expressed on the same construct or on a second, co-transfected vector. Various systems are used for selecting transfected cells, but usually include resistance to antibiotics, such as neomycin phosphotransferase, dihydrofolate reductase or DHFR, or glutamine synthetase. After transfection, cells are cultivated in medium having a selective agent. The cells that will survive are those that integrated the plasmid in their genome, including the gene that confers them drug resistance. After applying this selective pressure repetitively for some time, only the cells with a stable transfection will remain and can be cultivated further.
In RNAi-based gene silencing experiments the efficient delivery of siRNAs is a vital step. Synthetic siRNAs can be delivered by electroporation or by using lipophilic agents1. siRNAs have been used successfully to silence target genes; however, these approaches are limited by the transient nature of the response. The use of plasmid systems to express small hairpin RNAs helps overcomes this limitation by allowing stable suppression of target genes2.
4. RNA interference (RNAi)
RNAi is a process whereby short RNAs are used to modulate the expression of a target transcript, typically a protein-coding messenger RNA. Two types of small RNA molecules, microRNA (miRNA) and small interfering RNA (siRNA), are central to RNAi.
The RNAi process involves several steps. It is triggered by the presence in the cytoplasm of double-stranded RNA (dsRNA) that is either endogenously generated (through genomic transcription) or exogenously provided (by means of transfection). The dsRNA is recognized by an endonuclease (DICER), a member of the RNase III family which cleaves it into shorter dsRNAs, 21–23 nucleotides in length3,4. One of the two strands (the ‘guide’) is incorporated into a multi-protein complex known as RISC for RNA-induced silencing complex whereas the other strand (the ‘passenger’) is generally degraded. The short RNA that was incorporated into the RISC acts as a template that recognizes its “target,” typically a mRNA, in a sequence-dependent manner5,6. RISC acts on its target either by degrading it or by inhibiting its translation7–9. The end result is decreased abundance of the targeted molecule, and, in the case of protein-coding transcripts, decrease in the amount of protein product.
See also: RNAi – internal link
See also: RNAi – external link
5. Luciferase Assays
Luciferase is a generic term to refer to a class of oxidative enzymes involved in bioluminescence. The word derives from Lucifer, a Latin word meaning ‘light-bringing, morning star’ (lux, luc- ‘light’ + -fer ‘bearing.’). A well-known enzyme is the firefly luciferase from the firefly Photinus pyralis. “Firefly luciferase” as a laboratory reagent usually refers to the P. pyralis luciferase although recombinant luciferases from several other species of fireflies are also commercially available.
In luminescence reactions, light is produced by the oxidation of luciferin (a pigment):
The rates of this reaction between luciferin and oxygen are extremely slow until they are catalyzed by luciferase, sometimes mediated by the presence of cofactors such as calcium ions or ATP. The reaction catalyzed by firefly luciferase takes place in two steps:
Nearly all of the energy in the reaction is converted into photons. Photon emission can be detected by a light sensitive apparatus (e.g. luminometer) or modified optical microscopes. This allows observation of biological processes. Luciferase can be produced in the laboratory through genetic engineering for various purposes, and luciferase genes can be synthesized and inserted into organisms or transfected into cells. In biological research, luciferase is commonly used as a reporter to assess the transcriptional activity in cells that are transfected with a genetic construct containing the luciferase gene under the control of a promoter of interest.
Examples of applications that use luciferase include:
- monitoring of cell-signaling pathways
- RNAi: luciferase reporters can be used to quantitatively evaluate miRNA activity by inserting putative miRNA target sites downstream of the firefly luciferase gene.
- identification of proteins interacting in vivo using a system known as the two-hybrid system10. The potentially interacting proteins to be studied are brought together as fusion partners—one is fused with a specific DNA-binding domain, and the other protein is fused with a transcriptional activation domain. The interaction between two bona fide fusion partners is necessary for functional activation of a reporter gene driven by a basal promoter and the DNA motif recognized by the DNA-binding protein.
- monitoring of protein-protein interactions using bioluminescence resonance energy transfer (BRET), where two fusion proteins are made; one using the bioluminescent Renilla luciferase and the other fused to a fluorescent molecule. Physical interaction of the two fusion proteins results in energy transfer from the bioluminescent molecule to the fluorescent molecule, with a concomitant change from blue light to green light11.
- live-cell and in vivo imaging. Luciferase genes are commonly used as light-emitting reporters in cellular and animal models. Visualization of reporter expression using live-cell luciferase substrates allows nondestructive, quantitative assays and repeat measures of the same samples without perturbation.
6. Sequencing
Knowledge of DNA sequences has become indispensable for basic biological research, and in numerous applied fields such as diagnostic, biotechnology and biological systematics. The advent of DNA sequencing has significantly accelerated biological research and discovery. The rapid speed of sequencing attained with modern DNA sequencing technology was instrumental in the sequencing of the human genome as part of the Human Genome Project. When the project started in 1991, DNA sequencing entailed laborious radiation-based methods, with researchers manually loading electrophoresis gels and painstakingly reading bases from the resulting images. Today’s sequencing technologies are easier, faster and, more importantly, cheaper.
DNA sequencing includes several methods and technologies that are used for determining the order of the nucleotide bases—adenine, guanine, cytosine, and thymine—in a molecule of DNA.
6.1 Sanger sequencing
The first DNA sequences were obtained in the early 1970s by researchers using laborious methods. The chain-terminator (a.k.a. the “Sanger method” after its developer Frederick Sanger) rapidly became the method of choice. The classical Sanger method requires a single-stranded DNA template, a DNA primer, a DNA polymerase, normal deoxynucleotidetriphosphates (dNTPs), and modified nucleotides (dideoxyNTPs) that terminate DNA strand elongation. These ddNTPs will also be radioactively or fluorescently labeled for detection in automated sequencing machines.
The DNA sample is divided into four separate sequencing reactions, containing all four of the standard deoxynucleotides (dATP, dGTP, dCTP and dTTP) and the DNA polymerase. To each reaction is added only one of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP) which are the chain-terminating nucleotides, lacking a 3′-OH group required for the formation of a phosphodiester bond between two nucleotides, thus terminating DNA strand extension and resulting in DNA fragments of varying length.
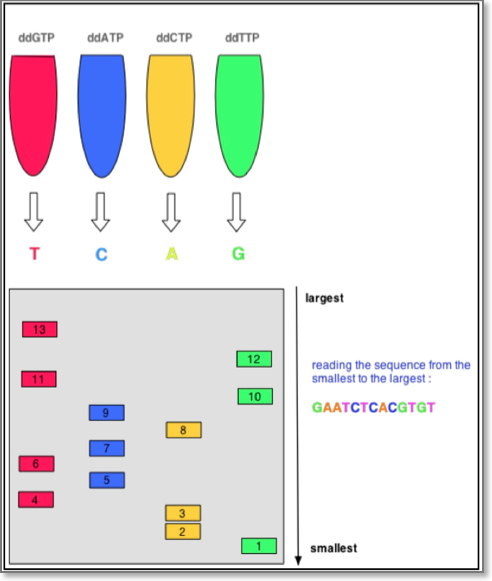 Sanger Chain Termination Sequencing
Sanger Chain Termination Sequencing
The newly synthesized and labeled DNA fragments are heat-denatured, and separated by size (with a resolution of just one nucleotide) by gel electrophoresis on a denaturing polyacrylamide-urea gel with each of the four reactions run in one of four individual lanes (lanes A, T, G, C); the DNA bands are then visualized by autoradiography or UV light, and the DNA sequence can be directly read off the X-ray film or gel image. The relative positions of the different bands among the four lanes are then used to read (from bottom to top) the DNA sequence.
Technical variations of chain-termination sequencing include tagging with nucleotides containing radioactive phosphorus for radiolabelling, or using a primer labeled at the 5’ end with a fluorescent dye. Dye-primer sequencing facilitates reading in an optical system for faster and more economical analysis, and automation. The later development by Leroy Hood and co-workers of fluorescently labeled ddNTPs and primers set the stage for automated, high-throughput DNA sequencing.
The automated Sanger method is considered a ‘first-generation’ technology, and newer methods are referred to as ‘next-generation sequencing’ methods. These newer technologies comprise various strategies that rely on a combination of template preparation, sequencing and imaging, and genome alignment and assembly methods.
See also: Sanger Chain termination sequencing – external link
See also: DNA Sequencing – The Sanger Method and Next generation sequencing – external link
6.2 Next generation sequencing
Next generation sequencing (NGS) has altered genomics research and allowed investigators worldwide to conduct experiments that were previously not technically feasible or affordable. The various technologies used continue to evolve with improvement in robustness and process streamlining, paving the way for translation into clinical diagnostics. The high-throughput capacity of NGS led to monumental accomplishments including the sequencing of entire genomes, from microbes to humans. All platforms used share a common feature: they do massively parallel sequencing of clonal or single DNA molecules that are spatially separated in a flow cell. This constitutes a paradigm shift from the Sanger method, which is based on the electrophoretic separation of chain-terminating products produced in individual sequencing reactions. NGS is performed by repeated cycles of polymerase-mediated nucleotide extensions or by iterative cycles of oligonucleotide ligation. NGS generates outputs measuring hundreds of millions to billions of nucleotides in a single instrument run, depending on the platform. The main providers of such platforms12 are:
Roche/454 Life sciences: The Roche/454 Life sciences sequencer is derived from the technological convergence of pyrosequencing and emulsion PCR and was the first to achieve commercial introduction in 2004. In pyrosequencing, each incorporation of a nucleotide by DNA polymerase results in the release of pyrophosphate, which initiates a series of downstream reactions that ultimately produce light by the firefly enzyme luciferase. The amount of light produced is proportional to the number of nucleotides incorporated. It can read 450 bases in a row and uses nucleotides that flash when the polymerase adds them to the DNA. Generally, this approach cannot properly interpret long stretches (>6) of the same nucleotide (homopolymer run) so these areas are prone to base insertion and deletion errors during base calling. By contrast, because each incorporation step is nucleotide specific, substitution errors are rare in Roche/454 sequence reads.
Watch: the 454 sequencing workflow – external link
Illumina/Solexa: Illumina/Solexa conceptualized an approach for sequencing single DNA molecules attached to microspheres. It can read 75-100 bases in a row and uses reversible dye terminators. The Illumina system utilizes a sequencing- by-synthesis approach: all four nucleotides are added simultaneously to the flow cell channels, along with DNA polymerase, for incorporation into the oligo-primed cluster fragments. In particular, the nucleotides are attached to a base-unique fluorescent label and the 3′-OH group is chemically blocked such that each incorporation is a unique event. An imaging step follows each base incorporation step. After each imaging step, the 3′ blocking group is chemically removed and each strand is prepared for the next incorporation by DNA polymerase. This series of steps continues for a specific number of cycles, as determined by the user, which permits discrete read lengths of 25–35 bases. A base-calling algorithm assigns sequences and associated quality values to each read and a quality-checking pipeline evaluates the Illumina data from each run, removing poor-quality sequences. A technical concern of Illumina sequencing is that base-call accuracy decreases with increasing read length.
Watch: the Genome Analyzer – external link
Live Technologies/Applied Biosystems: The SOLiD (Supported Oligonucleotide Ligation and Detection) System 2.0 platform, distributed by Applied Biosystems is a short-read sequencing technology based on ligation. It uses adapter-ligated fragment library similar to those of the other next-generation platforms, and uses an emulsion PCR approach with small magnetic beads to amplify the fragments for sequencing. SOLiD uses DNA ligase and a unique approach to sequence the amplified fragments, so that each template nucleotide is sequenced twice. This interrogation of each nucleotide base twice during independent ligation cycles yields a company- reported sequence consensus accuracy of 99.9% for a known target at 15-fold sequence coverage over sequence reads of 25 nucleotides.
Read: the SOLiD Sequencing Chemistry – external link
7. Next generation sequencing applications
7.1 DNA-seq
Whole genome and exome sequencing has provided us with important medical information embedded in individual genomes and exomes that can used into the practice of medicine for diagnostic purposes and therapeutic gains.
DNA sequencing has been used to elucidate the molecular genetic basis of susceptibility to common or even rare disorders and cancer. Some of these diseases are single-gene disorders whereas others encompass a familial aggregation of complex traits, such as coronary atherosclerosis. New insights into the pathogenesis of human disease can lead to the identification of new diagnostic tools, prognostic markers and drug targets13.
7.2 RNA-seq
NGS has enabled a powerful new approach, termed “RNA-seq,” for mapping and quantifying transcripts in biological samples. Total, ribosomal RNA–depleted, or poly(A)-enriched RNA is isolated and converted to cDNA. Although RNA-seq is in its early stages as a technology, it has already altered our view of the extent and complexity of eukaryotic transcriptomes and offers significant advantages over gene expression arrays. RNA-seq allows the characterization of the transcriptome without prior knowledge of the genomic sites of transcription origin, whereas arrays depend on tiling existing genomic sequences. RNA-seq is capable of single-base resolution and, compared with arrays, demonstrates a greater ability to distinguish RNA isoforms, determine allelic expression, and reveal sequence variants. The total number of reads that map to the exons of a gene, normalized by the length of exons that can be uniquely mapped defines the expression level of a given transcript. The dynamic range of RNA-seq for determining expression levels is at least ~5 orders of magnitude (at least ~17 PCR cycles), compared with ~3 orders of magnitude for expression arrays. In this context, RNA-seq has shown improved performance for the quantitative detection of both highly_produced transcripts and transcripts produced at low levels14.
7.3 ChIP-seq
Chromatin immunoprecipitation (ChIP) is a method used to identify the location of DNA binding sites on the genome for a particular protein of interest, such as transcription factors and histones. An antibody specific to a putative DNA binding protein is used and immunoprecipitation of the DNA-protein complex follows, resulting in the purification of protein-DNA complexes. Once the DNA fragments are separated from the complex, by reversing the cross-linking, they can be identified and quantified by PCR.
ChIP-sequencing, also known as ChIP-seq, is used to determine how transcription factors and other chromatin-associated proteins influence phenotype-affecting mechanisms. ChIP-seq combines ChIP with DNA sequencing to map the binding sites of DNA-associated proteins. This can be applied to the set of ChIP-able proteins and modifications, such as transcription factors, polymerases and transcriptional machinery, structural proteins, protein modifications, and DNA modifications. Determining how proteins interact with DNA is essential for fully understanding many cellular biological processes and disease states. This information is complementary to genotype and expression analysis.
7.4 RIP-seq
As mentioned above, mRNAs are subjected to multiple modifications and processing, many of which are regulated by RNA binding proteins (RBPs). The binding of RBPs to RNAs results in the formation of a ribonucleoprotein (RNP) complex to regulate many aspects of mRNA biogenesis and function. In order to gain a full understanding of RBPs role in cellular processes and disease, it is important to be able to accurately identify their targets and binding sites on a transcriptome-wide level. In recent years a variety of methods have been developed to identify RBP targets. Two of the most widely used are RNP immunoprecipitation (RIP) and ultraviolet (UV) crosslinking and immunopreciptiation (CLIP). However, these two methods need to be coupled to complex bioinformatic processes that will ultimately determine their success.
RNP immunoprecipitation (RIP) assay is a method that involves the purification of RNP complexes from cell extracts using specific antibodies to specifically recognize a protein of interest. Following the isolation of the RNP complex, the RBP and RNAs are dissociated from each other and high-throughput sequencing on the resulting mRNAs can be performed to identify the RBP-mRNA interactions.
References
- McManus, M.T., Petersen, C.P., Haines, B.B., Chen, J. & Sharp, P.A. Gene silencing using micro-RNA designed hairpins. RNA 8, 842-850 (2002)
- Dykxhoorn, D.M., Novina, C.D. & Sharp, P.A. Killing the messenger: short RNAs that silence gene expression. Nature reviews. Molecular cell biology 4, 457-467 (2003)
- Agrawal, N., et al. RNA interference: biology, mechanism, and applications. Microbiology and molecular biology reviews : MMBR 67, 657-685 (2003)
- Elbashir, S.M., Lendeckel, W. & Tuschl, T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes & development 15, 188-200 (2001)
- Hammond, S.M., Bernstein, E., Beach, D. & Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 404, 293-296 (2000)
- Bernstein, E., Caudy, A.A., Hammond, S.M. & Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409, 363-366 (2001)
- Elbashir, S.M., Martinez, J., Patkaniowska, A., Lendeckel, W. & Tuschl, T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. The EMBO journal 20, 6877-6888 (2001)
- Krol, J., Loedige, I. & Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nature reviews. Genetics 11, 597-610 (2010)
- Filipowicz, W., Bhattacharyya, S.N. & Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nature reviews. Genetics 9, 102-114 (2008)
- Fields, S. & Song, O. A novel genetic system to detect protein-protein interactions. Nature 340, 245-246 (1989)
- Angers, S., et al. Detection of beta 2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proceedings of the National Academy of Sciences of the United States of America 97, 3684-3689 (2000)
- Voelkerding, K.V., Dames, S.A. & Durtschi, J.D. Next-generation sequencing: from basic research to diagnostics. Clinical chemistry 55, 641-658 (2009)
- Marian, A.J. Medical DNA sequencing. Current opinion in cardiology 26, 175-180 (2011)
- Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature reviews. Genetics 10, 57-63 (2009)