Use the Off-Spotter web tool | Submit remote queries | Download Off-Spotter for local use
If you use any of the below please cite: Pliatsika, V, and Rigoutsos, I (2015) Off-Spotter: very fast and exhaustive enumeration of genomic lookalikes for designing CRISPR/Cas guide RNAs Biol. Direct 10(1):4
GENERAL
What is Off-Spotter
Off-Spotter is a tool designed by the Computational Medicine Center at the Sidney Kimmel Medical College of Thomas Jefferson University. It is designed to identify gRNAs followed by PAM on input and return ALL genomic locations that match a specific gRNA followed by a PAM sequence. For example if CATCATCAAAGACCAAAAGT and PAM=NGG is given, Off-Spotter will return ALL the sites on the human genome that CATCATCAAAGACCAAAAGTNGG exists together with all the sites that gRNAs different at mostly 5 bases exist, where N={A,C,G,T}. You can see the results for this example here. Multiple PAM sequences are available and the center is planning to add more in the future, see the results for the previous gRNA and PAM=NAG here. Additionally, the number of different bases that the input gRNA (mismatches) can be set to any number up to 5, see the initial example with up to 3 mismatches here. Finally, the seed region can be selected, see the initial example but with a fixed seed here.
A very useful option of Off-Spotter is the annotation display. When enabled all results will be annotated as to whether the off-target is located in a 5´ untranslated region (5´UTR), an amino acid coding sequence (CDS), a 3´UTR, a long non-coding RNA (lncRNA), etc. For each result that is located in one of the annotation regions, the gene ids, the transcripts ids and the common gene names are returned. Annotation is optional because it increases the running time, however, given Off-Spotter’s extreme performance it is effective to use.
Off-Spotter offers multiple display options such as column display and dynamic multi-sorting that enable the user to effectively select a suitable gRNA or design their own gRNA selection function.
Please see 1 for more information.
To access the web app click here.
WEB APP HELP
Input options and format
Genome
Off-Spotter currently operates on 4 genomes. Two human (hg19/GRCh37 and hg38/GRCh38), one mouse (mm10/GRCm38) and one yeast strain (w303)2. In order to select the genome of your choice click on the dropdown box on the app’s home page ![]() . We plan to add more species/genomes soon, check again or contact us for suggestions.
. We plan to add more species/genomes soon, check again or contact us for suggestions.
Annotation
Off-Spotter optionally annotates the results using ENSEMBL GRCh37.75. Currently the annotations include the following pre-mRNA (Unspliced), mRNA (5UTR, CDS, 3UTR), and lincRNA(Unspliced, Spliced). For each the gene IDs, the transcript IDs and the common gene names are returned. If a “-” is returned then this gRNA hits no known transcript or gene of this type. In order to get the annotations click the checkbox on the app’s home page![]() . To view part of the annotations see the column display options below. If you need more annotations you can use the source code locally or contact us.
. To view part of the annotations see the column display options below. If you need more annotations you can use the source code locally or contact us.
Mismatches
The number of mismatches define in how many different positions the hits returned will differ from the input gRNA. For example: CtTCATaAAAGtCCAAAAGT differs from CATCATCAAAGACCAAAAGT in exactly three positions. Those positions are indicated by small red letters. If you want to restrict some positions so that no mismatches will appear there, please see the “seed” option below. To set the maximum number of mismatches please select the dropdown box in the app’s home page ![]() .
.
Seed
Off-Spotter allows the user to FULLY define the seed size and location. To do so, please use the checkboxes on the input area of the home page 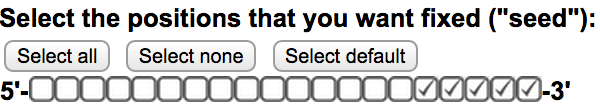 . Every checked box signifies that the corresponding location can not hold a mismatch. Thus only the same nucleotide that was given in the input in this location will be present on that location on all the results. This option decreases the number of results returned. Use the “select all”, “select none”, and “select default” buttons to set all, none and the default locations as seed.
. Every checked box signifies that the corresponding location can not hold a mismatch. Thus only the same nucleotide that was given in the input in this location will be present on that location on all the results. This option decreases the number of results returned. Use the “select all”, “select none”, and “select default” buttons to set all, none and the default locations as seed.
PAM
Off-Spotter currently includes the following 4 PAM sequences, NGG, NAG, NNNNACA, and NNGRRT (where R= A or T). More PAM sequences will be added in the future. To choose PAM, click on the dropdown box on the app’s home page ![]() .
.
Input format
The input can be either a genomic sequence up to 500 nucleotides in length OR one up to 20 gRNAs each on its own line. In the second case, please do not add the PAM sequence. For example the following two inputs are valid:
Genomic sequence:
CCTAGGAAGCATTCACATTTTCTTCTCGGGTCCCAGACGTGACTCACCA
Multiple 20mers separated by new lines and without PAM:
CATCATCAAAGACCAAAAGT
AGCTCTCTCGACGCAGGACC
You can explore more examples by clicking on the “Click here for sample” button under the input box.
Output options and layout
Output layout
The output page has 2 or 3 types of tables depending on the type of input.
The first table is located on the top left and contains a summary of the results found. There you can see:
- the gRNAs inserted/found on input and a link on their individual table
- the number of results per gRNA
- the strand on which the gRNA was found (if the input is a sequence)
- the reverse complement of the gRNAs found in the reverse strand (if the input is a sequence)
- the position where the gRNA was found on the input (if the input is a sequence)
- A histogram of the number of results per number of mismatches for each gRNA
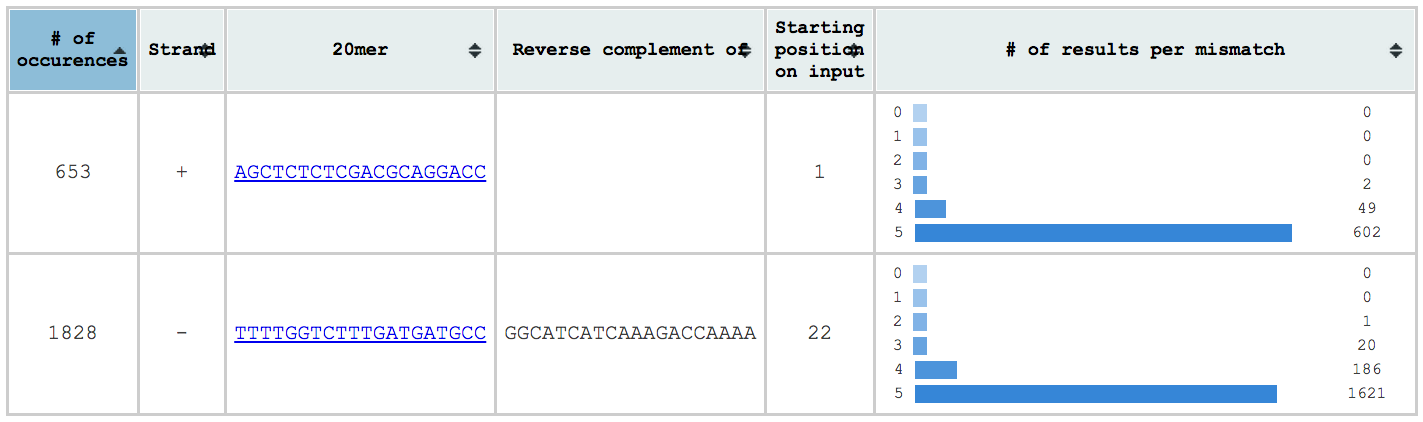
The second table is present only when a sequence is inserted and shows where on input each gRNA was found. The gRNAs are links to their individual and detailed tables.

The third is a collection of tables, one for each gRNA processed. Each of this tables contain all results that satisfy the input query and the following information for each of them:
- Chromosome name
- Strand
- gRNA start (including the PAM)
- gRNA end (including the PAM)
- gRNA processed
- gRNA found
- number of mismatches between the gRNA processed and the gRNA found
- Pre-mRNA (Unspliced) gene ids, transcript ids, and gene names
- mRNA (5UTR) gene ids, transcript ids, and gene names
- mRNA (CDS) gene ids, transcript ids, and gene names
- mRNA (3UTR) gene ids, transcript ids, and gene names
- lincRNA (Unspliced) gene ids, transcript ids, and gene names
- lincRNA (Spliced) gene ids, transcript ids, and gene names
- GC content in gRNA found (without the PAM)
- UCSC genome browser link for further visualization
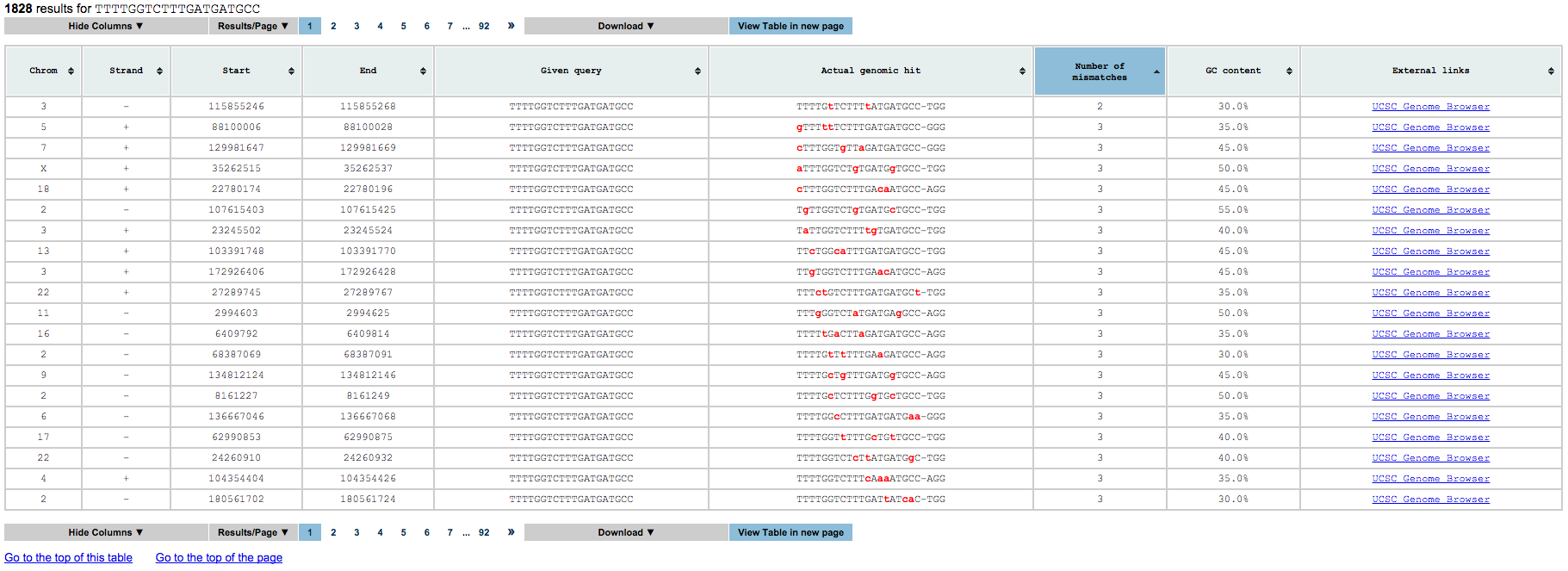
Navigating
There are several ways to navigate through the results. The top 2 tables contain links to the individual gRNA tables. Each individual gRNA table has links to the top of each table and to the top of the page. You can navigate through the results by sorting them, viewing part of them and by adjusting the number of results per pages.
Results per page
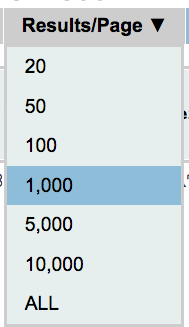 With this option you can select the number of results per page. The default value is 20 per gRNA. To select a different number of results per page move your mouse over the box shown on the side, select the number you prefer and right-click on it. Please keep in mind that only the results shown in the page can be sorted. Thus if you want to sort by a specific attribute you might need to select “ALL”.
With this option you can select the number of results per page. The default value is 20 per gRNA. To select a different number of results per page move your mouse over the box shown on the side, select the number you prefer and right-click on it. Please keep in mind that only the results shown in the page can be sorted. Thus if you want to sort by a specific attribute you might need to select “ALL”.
Column display
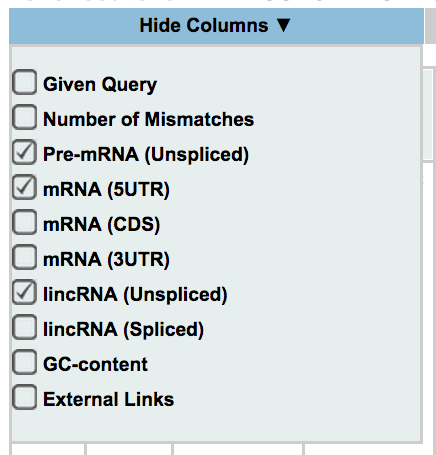 Off-Spotter allows the user to control which columns are displayed. By moving your mouse over the box shown on the side, and clicking on a checkbox you show/hide each column. The columns that are available for this option are the following: given query, number of mismatches, pre-mRNA (Unspliced), mRNA (5UTR, CDS, 3UTR), lincRNA(Unspliced, Spliced), GC-content, and external links.
Off-Spotter allows the user to control which columns are displayed. By moving your mouse over the box shown on the side, and clicking on a checkbox you show/hide each column. The columns that are available for this option are the following: given query, number of mismatches, pre-mRNA (Unspliced), mRNA (5UTR, CDS, 3UTR), lincRNA(Unspliced, Spliced), GC-content, and external links.
Sorting
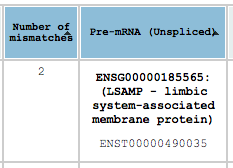 By clicking on the header of each column you can sort the results that are displayed in that page by that column. You can sort by multiple columns by holding the left-shift key. Note that only the results that are currently displayed on the page are sorted. If you want to sort through all the results the choose “ALL” from the results/page box.
By clicking on the header of each column you can sort the results that are displayed in that page by that column. You can sort by multiple columns by holding the left-shift key. Note that only the results that are currently displayed on the page are sorted. If you want to sort through all the results the choose “ALL” from the results/page box.
Download
 With the dropdown menu shown on the left you can download the hits of one gRNA (all pages) or all gRNAs inserted or found on the input (all pages of all tables). To do so, move your mouse on top of the box, select the option you want and right-click.
With the dropdown menu shown on the left you can download the hits of one gRNA (all pages) or all gRNAs inserted or found on the input (all pages of all tables). To do so, move your mouse on top of the box, select the option you want and right-click.
One-Table view
![]() If your results are too many, perhaps you prefer to see the tables setarately. Click on this button to view only a specific table on a new page. By clicking on the button again you can see all the results again.
If your results are too many, perhaps you prefer to see the tables setarately. Click on this button to view only a specific table on a new page. By clicking on the button again you can see all the results again.
SOURCE CODE HELP
This section contains information on how to use the source code. The code can be downloaded from this page.
How to download and run
To download visit the download page.
First time using a genome:
- Go to the table where all the Off-Spotter files are downloaded to.
- Run “make” to get the executables.
- Run “Table_Creation ” see below on Table creation section for details on <genome_file>. This creates 2 files called data.bin and index.bin. Those files must be on the same folder and the full path to that folder should be given as input to the next step.
- SHARED MEMORY:
- Run “Load_Memory -t <full path of .bin files> -g <hg19 or hg38 or mm10 or w303>”.
- Run “Results -i <input full path> -f -p <PAM sequence> -g <hg19 or hg38 or mm10 or w303> -n <max mismatch number> -a <Annotation file full paths divided by 2 dashes> -o <output file name>”. You can run multiple queries at the same time as long as the input and output files have different names.
- When you don’t want to run more queries, run “Detach_Memory -g <hg19 or hg38 or mm10 or w303>”.
MEMORY MAPPED FILES:
- Run “Results -i<input file> -f -p<PAM> -n<maximum mismatch number> -g<hg19 or hg38 or mm10 or w303> -t <full path to *.bin files> -m1 -a <annotation file list divided by –> -o <output file name>”. You can run multiple queries at the same time as long as the input and output files have different names.
If you already used this genome before and kept the .dat files, execute only step 4. You can find more details on each program and more options below.
General
The code of Off-Spotter consists of several separate programs. Depending on the memory options you will choose (see below) you’ll need to use 2 or 4 of them for Off-Spotter to run properly. Those programs are the following
- Off-Spotter_Table_Creation: This program creates two (2) tables and writes them in two (2) separate binary files. The first table is written to the data.bin file and contains all 20mers that are followed by any PAM on both strands of the input genome. The second is written in index.bin and contains the entries of a hash table on the entries of the previous table. This program needs to be run every time the user uses a new genome, otherwise the data.bin and index.bin files must be kept as they are used by the rest of the programs.
- Off-Spotter_Load_Memory: This program should be used only if you use shared memory and will load the .bin files on memory. Therefore it is necessary to have at least 23 GB of RAM available. You need to load the tables before you run the results program. However you don’t have to run this every time. You can load the tables and run Results as many times as you want without re-running this program. To remove the tables from memory you need to run Detach_Memory.
- Off-Spotter_Results: This program uses the loaded memory to generate the gRNA targets if you use shared memory. Or associates the files with shared memory and generates the gRNA targets if you use memory mapped files.
- Off-Spotter_Detach_Memory: This program should be used if you use shared memory only and it removes the tables loaded by Load_Memory from shared memory. Use when you don’t need to make more queries.
Memory Options
After version 0.2, Off-Spotter can run using shared memory or memory mapped files in order to parse through the files created by Table Creation. Shared memory is what Off-Spotter in the previous versions as well. The second option is recommended only for users who want to run Off-Spotter in parallel with other memory demanding processes. In general, shared memory is recommended as it is compatible with most systems and takes advantage the most of Off-Spotter’s extreme performance. Note that the memory requirement of 24 GB still holds in both cases as it’s necessary for Table Creation.
Depending on the memory option you select you will have to run a different subset of the programs. See the lists and the individual program information below.
Shared Memory
- Table Creation
- Load Memory
- Results (with memory option 0)
- Detach Table
Memory Mapped Files
- Table Creation
- Results (with memory option 1 and the path of the *.bin files)
Table Creation
Regardless of the memory option you’ll choose, you will have to use this program.
The available parameters are:
- -i : (Input) Genomic sequence file
- -o : (Output) Full path of output folder
An example invocation is the following:
./Table_Creation -i /home/user/me/hg19/full_genome.txt -o /home/user/me/hg19/Off-Spotter_bin/
Input
Table Creation accepts 2 arguments. The input file’s full path (-i) and the output folder’s full path (-o). The input file consists of the sequences of the chromosomes. There must be one file that holds all chromosomes. Before each chromosome there should be a header that starts with “>” followed by the chromosome number. The chromosome number can be followed by anything but another number. Each chromosome’s bases must be in one line. For example:
>1_hg19_DNA
NNNNNNNNNNNNNGGGG....GGGGNNNNNNNN
>2_hg19_DNA
NNNNNNNNNNNNNGGGG....GGGGNNNNNNNN
...
Output
The output of this program will be 2 binary files that contain all the information that Off-Spotter discovered for ALL PAMs, ALL gRNAs, both strands and ALL chromosomes. They will be located on the path you passed Table Creation (-o). Every time you run the “Results” program, Off-Spotter will search through those files and return the relevant results. You need to keep those files and pass them as parameters to the rest of the programs (see below). Also, because those files are depending on the input file you need to generate new for every genome you want to use. Note, you should not rename the files. If you want to use several genomes place the *.bin files on separate folders. Keep the files for every use of the same genome.
Load Memory
Use this program to load permanently the *.bin files in your memory. This way whenever you run “Results” the files will be available and you’ll get the most out of Off-Spotter’s extreme performance. The available parameters are:
- -g : (Genome) genome name
- -t : (Table) path to the table files
Note that you need to run this once for every genome you plan to use and that each takes roughly 23GB of memory.
An example invocation is:
./Load_memory -g hg19 -t /home/user/me/hg19/Off-Spotter_bin/
Input
The input of this program is the full path of the data.bin and index.bin files that were created by Table_Creation (-t) and the genome name (-g). The genome names that the code supports right now is hg19, hg38, mm10 and w303.
Output
None, this program will load the files in shared memory. You can check the contents of your memory by typing “ipcs”.
Results
This is the main program that you’ll use to get the genomic hits. The parameters you’ll use depend on your memory options. The available parameters are:
- -i : (INPUT) input gRNAs OR file that contains a sequence or gRNAs
- -f : (FILE) flag that indicates whether a file is used as an input – OPTIONAL
- -p : (PAM) PAM
- -n : (NUMBER) maximum number of mismatches
- -g : (GENOME) genome name
- -t : (TABLE) path to *.bin table files – OPTIONAL
- -o : (OUTPUT) write output to file – OPTIONAL
- -m : (MEMORY) memory option used – OPTIONAL (defaults to shared memory)
- -a : (ANNOTATION) full paths of annotation files – OPTIONAL
To use Off-Spotter with memory mapped files you must:
Include the parameters: -i, -p, -n, -g
Optionally use: -o, -f, -a, -m=0
An example is:
/oceanus/www/OffSpotterc++/bin/Results -iinput.txt -f -pG -n5 -ghg38 -a /home/hg38/classfiles/Unspliced--/home/hg38/classfiles/5UTR--/home/hg38/classfiles/CDS--/home/hg38/classfiles/3UTR
To use Off-Spotter with memory mapped files you must:
Include the parameters: -i, -p, -n, -g, -m=1, -t
Optionally use: -o, -f, -a
An example invocation is:
/oceanus/www/OffSpotterc++/bin/Results -iCATCATCAAAGACCAAAAGT -pG -n5 -ghg38 -t/oceanus/www/OffSpotterc++/hg38/ -m1 -a /home/hg38/classfiles/Unspliced--/home/hg38/classfiles/5UTR--/home/hg38/classfiles/CDS--/home/hg38/classfiles/3UTR
Input
Input and Input File (-i, -f)
The input file can have 2 formats. It can be a list of gRNAs or a filename. If it is a filename you need to use the -f parameter too to indicate that.
List of gRNAs
The list can be as long as you want but remember that the running time depends on the number of gRNAs as well as the number of mismatches and the number of hits of each gRNA. The gRNAs must be divided by — and shouldn’t have any spaces among them. An example list of gRNAs is:
-i CATCATCAAAGACCAAAAGT--TCTAACACATAAGGAGCATG
Input file
If you want to find the gRNAs of a sequence and their genomic hits, the input file must be in FASTA format. The headers and the empty lines will be ignored. At least one line must have length more than 20 or everything is considered a list of gRNAs.
If you want to enter the gRNAs directly then the input file must have only gRNAs and only one per line. Thus the file has 20 characters per line.
PAM (-p)
The available PAMs are NGG, NAG, NNNNACA, NNGRRT.
Maximum mismatch number (-n)
An integer between 0 and 5 inclusive. If you use x then you’ll get all results that have at most x different bases than the input. Hence, 0 returns exact hits only, 1 returns exact hits and hits that differ in 1 position out of the 20 etc. The N’s in the PAMs are not considered as mismatches, no need to take them into account.
Genome name (-g)
The available genome names are hg19, hg38, mm10 and w303.
File output (-o – OPTIONAL)
If this parameter is used and followed by a filename the output will be written to the file otherwise will be printed on the standard output.
Memory Option (-m)
Use 0 for shared memory or 1 for shared memory files. You can omit the parameter if using shared memory.
Path to *.bin table files (-t)
This parameter should be added if -m is 1, in other words if you are using memory mapped files. It should be followed by the path of the *.bin files created by Table Creation.
Annotation file (-a optional)
The annotation file should be ordered and also should have the following structure: chromosome name t strand t start position t end position t info where chromosome is a number and X=23, Y=24, MT=25. And strand is + or -. For example:
1 - 739121 739137 ENSG00000269831_ENST00000599533_AL669831.1_Uncharacterized protein
you can give multiple files but you should give the full path of each and separate them with “- -” and no spaces.
Output
The output has one line per hit. Each line has the following format:
- Chromosome name
- Strand
- Coordinate start
- Coordinate end
- gRNA used for search
- genomic hit with PAM resolved
- number of mismatches between gRNA used and genomic hit
- Annotation info or – if no entry exists in the annotation files for those coordinates. That column will appear as many times as the annotation files entered, once per file.
Detach Memory
Use this program to detach the permanently loaded memory if using shared memory. The only available parameter is:
- -g : (Genome) genome name
Note that you need to run this once for every genome you loaded.
Input
The genome name.
Output
None.
Common errors
Here’s a list of common errors that have been reported to us. If you have encountered a new error, please contact us.
- Although I have a lot of memory, I get “No space left on device” on Load_Memory. That problem is usually encountered when /proc/sys/kernel/shmmax, /proc/sys/kernel/shmmni and, /proc/sys/kernel/shmall don’t allow heavy shared memory usage. shmmax is the max size (bytes) that you are allowed to load on shared memory. It has to be larger than the total space of shared memory you are going to use. shmmni is the total number of shared memory segments you can have. That’s usually 4096 which is enough in most case. Finally shmall, is the number of pages you can have on shared memory. Set this to, at least, shmmax/Page size of your system. Also, confirm that you’ve removed shared memory segments that are not currently used with Detach_Table. You can see the tables/segments that are currently on shared memory by using “ipcs”.
Code version and changelog
Current version: v0.2.2 RELEASED 10/26/2015
- Added library for compatibility with latest gcc version
- Improved error printing in Load_memory
v0.2.1 RELEASED 8/6/2015
- Added makefile
- Handled warnings that haven’t changed the functionality/correctness of the previous version
v0.2 RELEASED 4/30/2015
- Added handling of newest human genome, newest mouse genome and the w303 strain of yeast
- Included memory mapped files as a memory option
- Added parameter for input. Input can be either file that contains a list of gRNAs or a sequence, or list of gRNAs
- Added parameter for output. Output can be written to file or printed in stdout
- Improved parameter passing and error checking
v0.1.1 RELEASED 2/6/2015
- Initial version
References
- Pliatsika, V, and Rigoutsos, I (2015) Off-Spotter: very fast and exhaustive enumeration of genomic lookalikes for designing CRISPR/Cas guide RNAs Biol. Direct 10(1):4
- Ralser, M, Kuhl, H, Ralser, M, Werber, M, Lehrach, H, Breitenbach, M, & Timmermann, B (2012) The Saccharomyces cerevisiae W303-K6001 cross-platform genome sequence: insights into ancestry and physiology of a laboratory mutt Open Biology 2(8):120093
You are visitor number 7651.